
The FDA regulates 25% of American consumer spending. And perhaps more importantly, the FDA regulates some of the most critical goods sold in our economy, such as life-saving drugs and medical devices. Delaying the approval of life-saving drugs and medical devices, and discouraging their development through those delays, costs lives.
Unfortunately, the FDA has been a major roadblock for medical innovation, through its delays and bureaucratic requirements. It costs about $2.6 billion to get a new drug approved by the FDA, according to a Tufts study. Contrast that with the largely unregulated world of computers and software, which has become synonymous with technology, where within a generation, we now have personal computers, tablets, and smartphones. What does the medical industry have to show for that same time period?
To a large extent, the lack of progress is because the government is strangling innovation. And that can soon change if Donald Trump nominates Jim O’Neill to lead the Food and Drug Commission (FDA).
O’Neill met with Trump last week to discuss becoming FDA commissioner. He’s a passionate advocate of finding new ways for the government to spur more innovation in medicine. If nominated and confirmed, he would make the FDA an ally of innovation instead of an obstacle.
O’Neill is currently managing director of Mithril Capital Management, a venture capital firm co-founded by Peter Thiel. Before working for Thiel, he spent six years working in senior roles at the Department of Health and Human Services under President George W. Bush, and worked closely with the FDA.
While Apple and Google outgrew their garages into multibillion-dollar companies, the government stood in the way of any aspiring Steve Jobses and Mark Zuckerbergs in the medical industry that wanted to get their innovations to market and improve on them. In fact, not only is over regulation suppressing health care startups, it’s also scaring off some of our best and brightest companies. Sergei Brin, Google co-founder, stated to a group of tech CEOs, “Health is just so heavily regulated,” “it’s just a painful business to be in.”
The FDA was built for a world where most medical innovations came from large, multinational corporations that had the resources necessary to undergo expensive, extensive clinical trials. But it’s long past time for the FDA to adapt to a world where innovation can come from both large pharmaceutical companies and startups alike. New firms are a key source of competition and innovation, and the FDA needs to nurture those new firms instead of stifling them.
The FDA’s slow-moving bureaucracy has been obstructive for the rapid medical and drug innovation coming out of Silicon Valley. Take, for example, 23andMe, a revolutionary company which offered disease risk profiles based on genetic information. The FDA prevented 23andMe from offering information about disease risk profiles. Instead, 23andMe has focused on providing ancestral information. Recently the FDA approved two specific product offerings, but the era of personalized medicine is delayed.
Another example of how the FDA has failed to adapt to the startup era is their pre-submission program. The pre-submission program allows companies to get informal feedback on their ideas before conducting pre-clinical trials. However, even getting informal written feedback takes 75 to 90 days, a lifetime for an early stage startup. With more feedback from the FDA, more ideas would be brought before investors and more new drugs could be developed.
Last May, the Wall Street Journal reported that tech-savvy families were building their own automatic insulin pumps. The FDA was so slow in approving insulin pumps that 50 or more families simply built their own. Millions of sufferers of diabetes were subjected to unnecessary hardship because of an overly onerous regulatory process. Luckily, several months later the FDA did approve an automatic insulin pump.
The FDA is required to find that new drugs are both safe and effective before approving them. The 1962 Kefauver-Harris Amendments added the efficacy requirement for new drugs. Sam Peltzman, in the Journal of Political Economy, estimated the effect of the new requirements on the number of new drugs approved by the FDA. The figure below shows the rapid drop in the approval of new drugs, labeled here as new chemical entities (NCEs):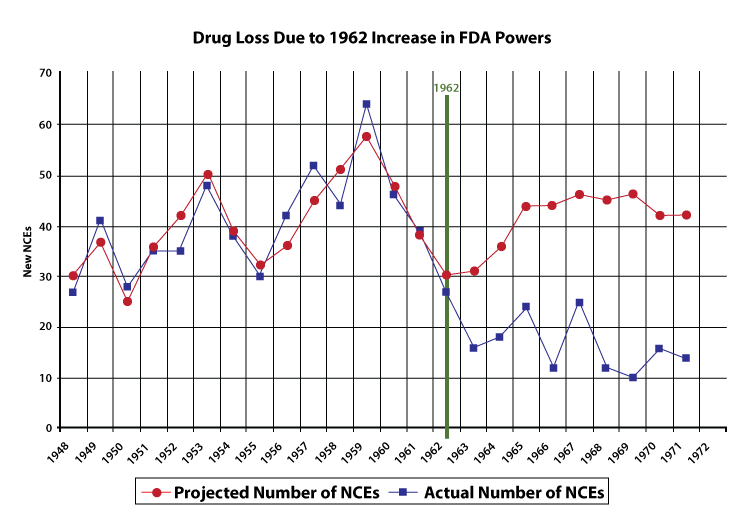
This is not because drugs that were otherwise ineffective were not being approved. A subsequent literature review in 1978, concluded: “The hypothesis that the observed decline in new product introductions has largely been concentrated in marginal or ineffective drugs is not generally supported by empirical analyses.” It’s possible that instead, fewer drugs were being developed in the first place.
O’Neill represents the best candidate to bring the FDA into the 21st century, into the age of startups, personalized medicine, and the rapid development of new drugs. He understands both the FDA as well as biotech startups. This gives him unique insight into how to reform the FDA, ensuring the FDA can be a partner, rather than a roadblock, for innovation. If the next 25 years can be as revolutionary for medicine as the past 25 years have been for computers, that would be tremendous, and O’Neill is determined to help us get there.
Follow Mark on Twitter.








